The host cell infection by HIV starts by the entry of the virus into the cell. The major target cells of HIV are CD4+ T lymphocytes, which also express chemokine receptors CCR5 and CXCR4 that are exploited by HIV to enter the cells, hence also called HIV co-receptors. The virus entry starts with the adhesion of the virion to the target cell and ends with the fusion of the cell and viral membranes followed by delivery of the viral core into the cytoplasm (reviewed in Wilen, C. B, Tilton, J. C, & Doms, R. W. 2012). Attachment of the virion (fig. 1(1)) can be relatively nonspecific, for instance, HIV envelope protein (Env, the trimer of gp120 and gp41 heterodimers, where gp41 initially is hidden) can interact with negatively charged cell-surface heparan sulfate proteoglycans (Saphire, A. C, Bobardt, M. D, Zhang, Z, David, G, & Gallay, P. A. 2001). More specific adhesion includes interactions between Env and a4b7 integrin (Arthos, J, Cicala, C, Martinelli, E, Macleod et al., 2008; Cicala, C, Martinelli, E, McNally,et al., 2009) or CLRs such as DC-SIGN (Geijtenbeek, T. B, Kwon, D. S, Torensma et al., 2000). Either way of adhesion most likely brings Env into close proximity with the host CD4 and a coreceptor, leading to the fusion of viral and target cell membranes (fig. 1.) (Orloff, G. M, Orloff, S. L, Kennedy, M. S, Maddon, P. J, & McDougal, J. S. 1991).
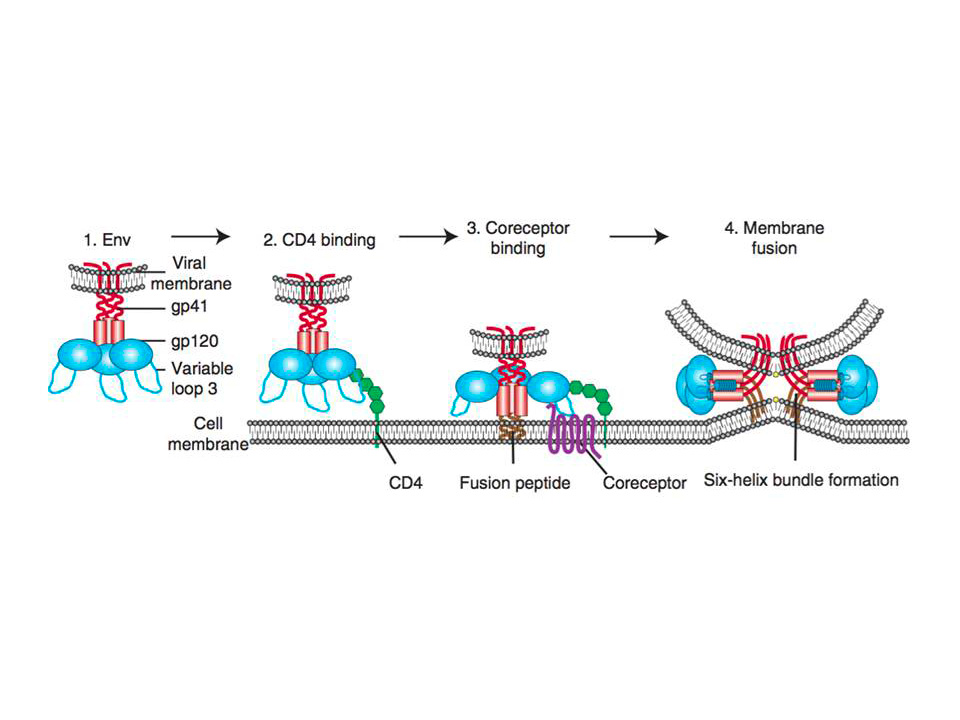
Figure 1 : Schematic representation of HIV entry to the target cell.
(1) HIV first attaches to the host cell via Env binding to host cell surface structures. Then binding to CD4 receptor
occurs (2), which causes conformational changes in Env, allowing coreceptor binding, which is mediated in part by the V3 loop of Env (3). Binding to CD4 also exposes gp41 subunit and thus initiates the membrane fusion process as the fusion peptide of gp41 inserts into the target membrane, followed by six-helix bundle formation and complete membrane fusion (4)
Env binding to its primary receptor CD4 (fig. 1(2)), a member of the immunoglobulin superfamily that enhances T-cell receptor (TCR)-mediated signaling, is absolutely required for the infection. This binding event induces rearrangements in gp120 subunit of Env, which ultimately result in V3 loop repositioning and bridging sheet exposure that are essential for coreceptor engagement (reviewed in Wilen, C. B, Tilton, J. C, & Doms, R. W. 2012). Subsequent binding to the coreceptor (fig. 1(3)), CCR5 or CXCR4 depending on the virus strain R5 or R4, triggers the membrane fusion potential of Env, and usually is followed by the “surfing” of the virus particle to the site where productive membrane fusion may occur (Sherer, N. M, Jin, J, & Mothes, W. 2010). It is thought that HIV might usurp the host cell machinery to reach cell surface sites where membrane fusion can occur. Besides, HIV may need to be endocytosed by the host cell for productive membrane fusion to occur (Miyauchi, K, Kim, Y, Latinovic, O, Morozov, V, & Melikyan, G. B. 2009).
Coreceptor-bound Env undergoes conformational changes that expose the hydrophobic gp41 fusion peptide (fig. 1(3)), which then inserts into the host cell membrane and folds to form six helix bundle (fig. 1(4)). The latter is the driving force that brings the opposing membranes into close proximity, resulting in the formation of a fusion pore (reviewed in Melikyan, G. B. 2008).
Once the virus enters the cell, it can start its replication and productive infection, which ultimately lead to the depletion of CD4+ T lymphocytes in the body, and thus the acquired immunodeficiency syndrome (AIDS).
Although the major target of HIV is CD4+ T cells, earlier studies have shown that DCs are crucial for HIV-1 infection enhancement and dissemination in mucosa, in the case of sexual HIV transmission, which leads to the productive infection of the CD4+ T cells and the burst of the disease (Cameron et al., 1992 Pope, M, Betjes, M. G, Romani, N. et al., 1994).