Mono-TACA vaccines have shown promising results in early clinical trials, nevertheless this approach does not take account of the inherent heterogeneity of TACAs expressed on transformed cell surface. Danishefsky, Livingston, and co-workers hypothesize that the combination of different carbohydrate antigens, associated with a particular cancer type, could be beneficial for the induction of a more potent immune response, reducing the ratio of tumor cells that can undergo immunoevasion. The initially envisaged strategy consisted in the administration of pooled mono-TACA KLH-based conjugate vaccines, containing up to seven different TACAs.ragupathi et al., 2002 ragupathi et al., 2003 Although this approach enabled the production of antibody titers against each individual antigen (to a similar extent, compared to Abs obtained through immunization with single TACAs), several fundamental limitations discouraged this route. Firstly, the pooled mono-TACA vaccine approach requires the use of increased levels of carrier protein, which could be detrimental and may contribute to variable clinical responses. oyelaran & gildersleeve, 2010 Consequently, the vaccine mixture would require the validation of each monomeric component for its clinical application. Finally, due to the low-yielding antigen-protein conjugation step, this route results not so efficient from a synthetic point of view. oyelaran ragupathi ragupathi
The intriguing idea of developing hetero-TACA-based anticancer vaccines required strong efforts for the chemical synthesis of key building blocks, namely glycosylamino acids, which have been exploited in the cassette assembly, described by Danishefsky and co-workers. fernández-tejada et al., 2015. As described in Scheme 2, the first generation of semi-synthetic mono-TACA conjugated vaccines were obtained by reductive amination between the aldehyde group on the carbohydrate moiety, obtained through ozonolysis of the terminal allyl group, and lysine amino group of KLH. The allyl glycoside was achieved through epoxidation of the glycal moiety, as depicted in scheme 5.

This strategy presents some disadvantages due to the instability of the allyl moiety towards the conditions required for the global deprotection, which require its introduction at the late per-acetate stage of the carbohydrate synthesis. Moreover, the glycal epoxidation step shows moderate stereoselectivity, causing a loss of material at a late stage of the synthetic pathway. Nevertheless, this protocol has constituted the basis for the development of three methods which has served for reaching TACA-based glycosylamino acids (Scheme 6).
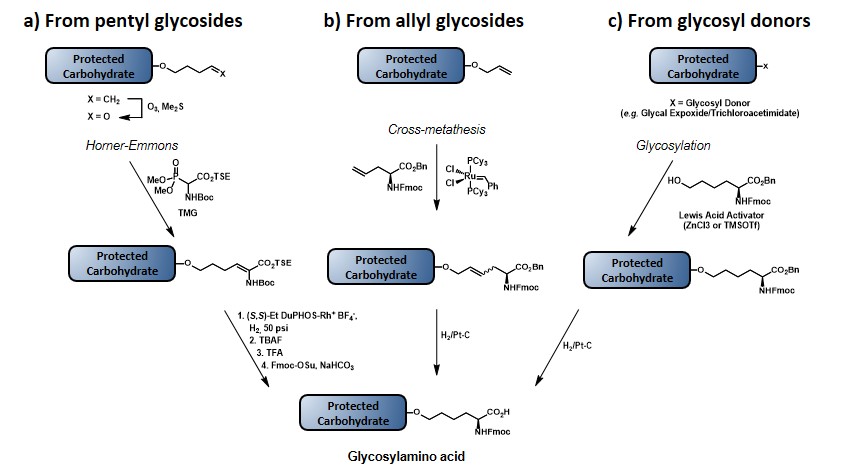
The first method (Scheme 6a) involves the utilization of the more stable pentenyl glycoside, which allows for the glycal functionalization at a much earlier stage compared to the allyl ether protocol, thus reducing the impact of the chemical loss associated to the glycal epoxidation step. After ozonolysis of the terminal olefin, the resulting aldehyde is reacted under Horner-Emmons conditions, by using a Boc- and TSE-protected phosphonate, in the presence of 1,1,3,3-tetramethylguanidine (TMG) as a base to give a dehydroamino acid, as shown. Subsequent enantioselective reduction of the double bond by means of an enantiopure phospholane, namely 1,2-bis((2S,5S)-2,5-diethylphospholano)ethane(cyclooctadiene)rhodium(I) tetrafluoroborate, followed by protecting group manipulation, leads to the desired glycosylamino acid. (allen, harris & danishefsky, 2001) The second method (Scheme 6b) starts with the olefin cross-metathesis between protected allyl glycoside and allyl glycine in the presence of ruthenium catalyst. Reduction of the resulting olefin, with concomitant benzyl ester deprotection provides the glycosylamino acid building block. Despite the drawbacks related to the allyl glycoside synthesis (vide supra), this route exploits the commercially available enantiopure form of allyl glycine, with no need to introduce a stereocenter.(biswas, coltart & danishefsky, 2002) In the third method (Scheme 6c) the amino acid functionality is directly introduced by coupling the glycal epoxide, or the trichloroacetimidate donor with hydroxynorleucine in the presence of a Lewis acid. Although this method allows for the direct introduction of the amino acid moiety, its efficiency is still limited by the need to synthesize hydroxynorleucine and the moderate stereoselectivity of the glycal epoxidation. Despite their limitations, each of these three methods has been used to obtain TACA-based glycosylamino acid, where the carbohydrate moiety is separated from the amino acid stereocenter by a four- or six-carbon linker. In earlier reports a trivalent-hetero-TACA vaccine, incorporating globo-H, Ley and Tn antigens gave a proof of concept by allowing the production of antibodies against the three antigens, which strongly reacted with the MCF-7 breast cancer cell line (32, Figure 5).(ragupathi et al., 2002) The first generation of pentavalent-hetero-TACA vaccine, comprising globo-H, Ley, STn, TF and Tn antigens has proved its effectiveness compared to its relative pooled monomeric formulation; nevertheless, only a minimal response to the Ley antigen was observed (33, Figure 5).(ragupathi et al., 2006) The authors suggested that the low response was due to the nature of Ley, as its high endogenous expression levels make this antigen less effective, compared to those that are naturally present only at low levels. In 2009, the Danishefsky group reported the synthesis and preclinical data of a second-generation pentavalent-hetero-TACA vaccine, where the Ley antigen was replaced with GM2 antigen (34, Figure 5).(zhu et al., 2009)
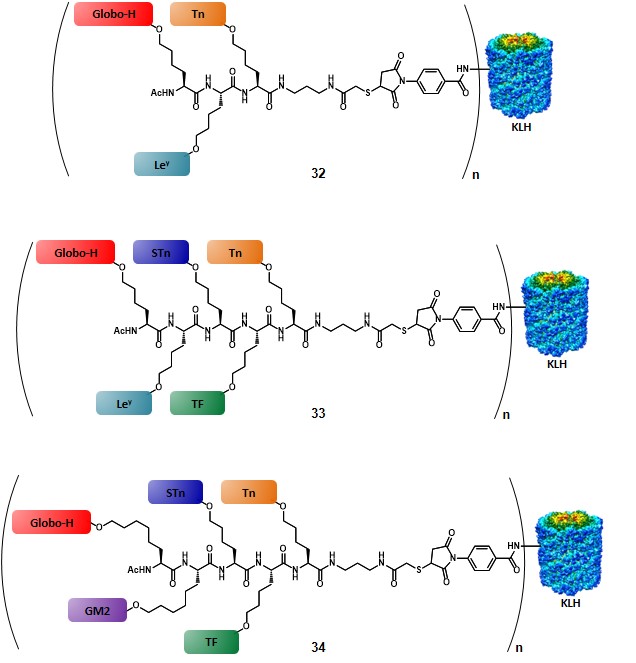
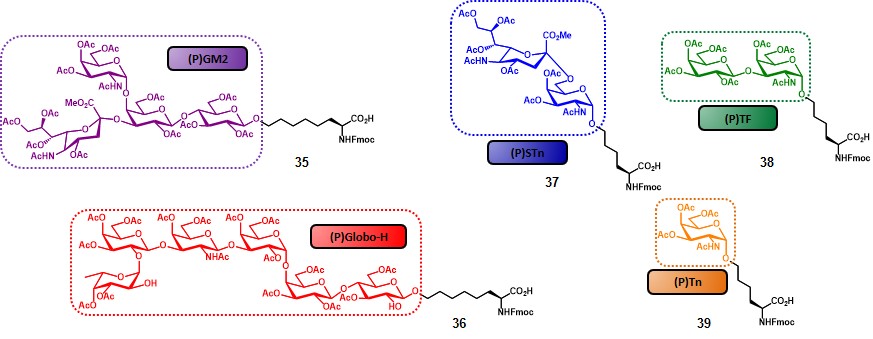
The choice of replacing Ley antigen with the tetra-saccharide GM2 was based not only on its presence in several cancer types (Table 2), but also taking into account previous studies, showing that (i) GM2-induced antibodies are reactive against human GM2-positive cells, and (ii) that a correlation between those antibody levels and survival rate could be traced.(livingston et al., 1987) The first step for the synthesis of the complicated “unimolecular pentavalent vaccine” involved the preparation of the glycosylamino acids 35-39 (Figure 6), by using the procedures described in scheme 6. In order to minimize steric hindrance phenomena, compounds 35 and 36 were synthesized with a six-carbon spacer. These variants were obtained through the cross-metathesis procedure (Scheme 6b) by starting from pentyl glycosides. The rationale of the designed synthetic route involved the positioning of the TACAs from the smallest (i.e. Tn) to the largest (i.e. globo-H) through a sequential assembly, with the aim to reduce the loss of the more complex antigens by introducing them at a later stage of the synthesis (Scheme 7).
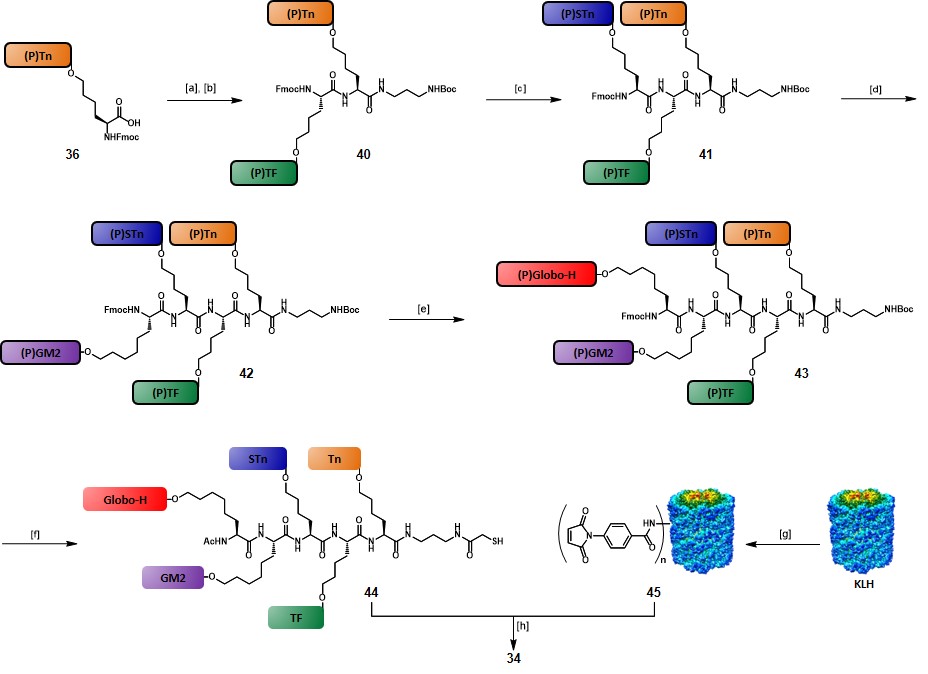
The synthetic route to 34 started with the coupling of the tert-butyl-N-(3-aminopropyl)carbamate linker on cassette glycosylamino acid 36. Subsequent iterative Fmoc-removals and glycosylamino acid couplings, involving 38, 37, 35 and 36, respectively, afforded fully protected intermediate 43, with a 57% yield over nine steps. The Fmoc protecting group of 43 was cleaved and substituted with an acetate, then Boc-deprotection and functionalization of the resulting free amino group with a S-acetylthioglycolic activated ester, followed by global deprotection under Zemplén conditions, gave the “unimolecular pentavalent glycopeptide” 44 with a 68% yield over five steps. Another objective of the authors was to increase the protein-conjugation efficiency. Therefore, they adopted a slightly modified procedure compared to their precedents works (See scheme 4), in which KLH protein was reacted with m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester (Sulfo-MBS) in pH 6.0 phosphate buffer for 1 hour, to give pure maleimidated KLH 45 after G25 Sephadex column purification. Freshly prepared glycopeptide 44 was passed through TCEP gel immediately prior to use, in order to minimize the amount of the putative disulfide oxidation by-product. Then, conjugation between 44 and 45 was carried out in PBS at pH 6.5–7.0. After 4 hours reaction and removal of unreacted 44 by a 30·000 M.W. cut-off filter, pentavalent-hetero-TACA vaccine 34 was obtained in 50% yield, and the recovery yield was approximately 98%. By using this modified procedure the authors achieved a more efficient glycopeptide: KLH conjugation ratio of 505:1, compared to the 228:1 ratio obtained for vaccine 33 (Figure 5). Five female mice (C57BL/6J) were immunized with vaccine 34 (10 µg) plus QS-21 (20 µg) and the collected sera, along with pre-immunization sera, underwent ELISA assays to assess IgM and IgG titers associated with each of the five TACAs. While none of the pre-vaccination sera showed reactivity against the five antigens, post-immunization sera contained substantial titers of antibodies, which proved to be reactive against each of the five TACAs, thus indicating that the immunological properties of the single TACAs were conserved when integrated into this complex vaccine construct.
(livingston zhu ragupathi ragupathi biswas, allen, fernández-tejada)